nf-core/rnafusion
RNA-seq analysis pipeline for detection of gene-fusions
1.0). The latest
stable release is
4.0.0
.

![]()

![]()

![]()
nfcore/rnafusion uses RNA-seq data to detect fusions genes.
The workflow processes RNA-sequencing data from FastQ files. It runs quality control on the raw data (FastQC), detects fusion genes (STAR-Fusion, Fusioncatcher, Ericscript, Pizzly, Squid), gathers information (FusionGDB), visualizes the fusions (FusionInspector), performs quality-control on the results (MultiQC) and finally generates custom summary report.
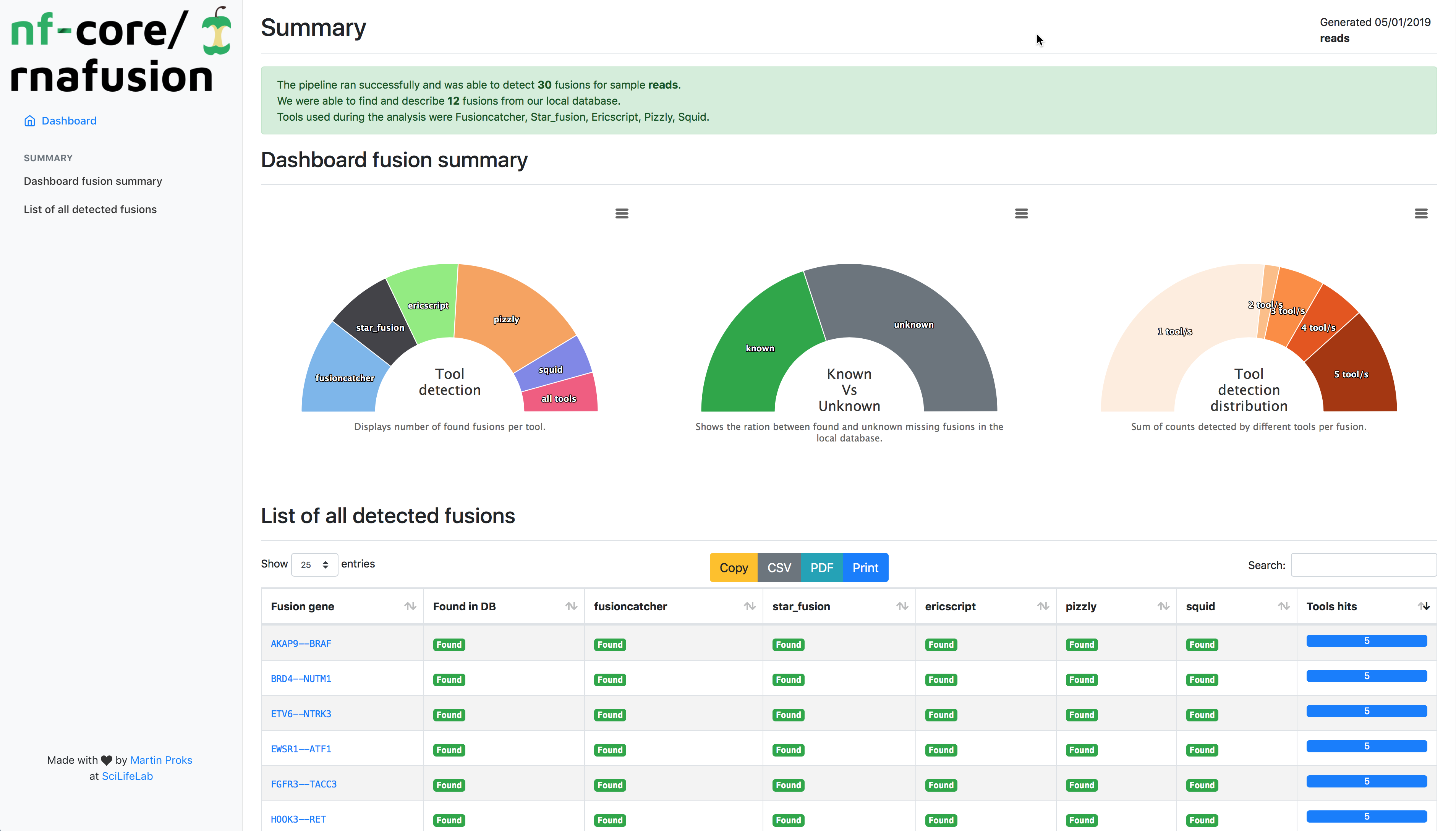
The pipeline works with both single-end and paired-end data, though not all fusion detection tools work with single-end data (Ericscript, Pizzly, Squid and FusionInspector).
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker / singularity containers making installation trivial and results highly reproducible.
| Tool | Single-end reads | CPU (recommended) | RAM (recommended) |
|---|---|---|---|
| Star-Fusion | Yes | >=16 cores | ~30GB |
| Fusioncatcher | Yes | >=16 cores | ~60GB |
| Ericscript | No | >=16 cores | ~30GB |
| Pizzly | No | >=16 cores | ~30GB |
| Squid | No | >=16 cores | ~30GB |
| FusionInspector | No | >=16 cores | ~30GB |
TL;DR: Make sure to download all required references for each tool. More details can be found in section tools.
nextflow run nf-core/rnafusion --reads '*_R{1,2}.fastq.gz' --genome GRCh38 -profile docker --star_fusion --fusioncatcher --ericscript --pizzly --squid --fusion_inspectorFor available parameters or help run:
nextflow run nf-core/rnafusion --helpDocumentation
The nf-core/rnafusion pipeline comes with documentation about the pipeline, found in the docs/ directory:
- Installation
- Pipeline configuration
- Running the pipeline
- Output and how to interpret the results
- Troubleshooting
Use predefined configuration for desired Institution cluster provided at nfcore/config repository.
run with
video introduction
subscribers
stars
open issues
open PRs
last release
last update
included modules
included subworkflows
contributors
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")