We are thrilled to announce the first release of the nf-core/fastquorum pipeline, which implements the fgbio Best Practices FASTQ to Consensus Pipeline to produce consensus reads using unique molecular indexes/barcodes (UMIs).
Developed by Nils Homer at Fulcrum Genomics, the pipeline utilizes the fgbio Bioinformatics toolkit to enable producing ultra-accurate reads for low frequency variant detection in Genomics/DNA Sequencing.
But Why?

As described in Salk et al 2018, finding the one in a thousand frequency variant, or even the one in the million frequency variant, is extremely important across a diversity of applications, including but not limited to:
- Cancer (ctDNA)
- Prenatal diagnosis (cffDNA)
- Mutagenesis
- Aging
- Antimicrobial Resistance
- Forensics
Sequencing Errors Obscure the Truth

Sequencing error, and errors from the library preparation process itself, makes it extremely difficult to obtain this accuracy at such incredible resolution.
Molecular Barcoding to the Rescue

Obtaining such high accuracy has been achieved through molecular barcoding that enables squashing random error through multiple observations of a single DNA source molecule. These molecular barcodes are commonly referred to as Unique Molecular Indexes, or UMIs. They are attached to a DNA source molecule to uniquely identify it. After amplification, the multiple observations can be compared to vote on a consensus. Or form a quorum, if you will.

Molecular barcodes can be added at various points in the library preparation process, including prior to or during amplification, for a single strand or to both strands of a double-stranded duplex molecule. In particular, Duplex Sequencing can be used to identify reads from both strands of a double-stranded source molecule, squashing strand-specific errors that can occur during library preparation (e.g. PCR errors, oxidative damage due to hybrid capture).
This means UMIs can be found in the index/sample-barcoding reads (i7/i5 reads in Illumina parlance), or inline in the genomic/template reads themselves (R1/R2). Furthermore, there sometimes exists extra “spacer” sequence which should be removed prior to alignment and downstream analysis.

In fgbio, as well as in fqtk, sgdemux, and also in Picard, Read Structures are used to describe how the bases in a sequencing run should be allocated into logical reads.
It serves a similar purpose to the --use-bases-mask in Illumina’s bcltofastq software, but provides some additional capabilities.
The following handful of examples from the fgbio wiki demonstrate the recommended way to describe a sequencing run in two different ways.
Firstly as a single Read Structure for the entire run as you might use with picard IlluminaBasecallsToSam, and secondly as a set of Read Structures that would map one-to-one with the physical reads after fastq-conversion and optionally adapter trimming (which will create variable length reads).
A few examples:
- A simple 2x150bp paired end run with no sample or molecular indices:
150T150T[+T, +T]
- A 2x75bp paired end run with an 8bp I1 index read:
75T8B75T[+T, 8B, +T]
- A 2x150bp paired end run with an 8bp I1 index read and an inline 6bp UMI in read 1:
8M142T8B150T[8M+T, 8B, +T]
- A 2x150bp duplex sequencing run with dual sample-barcoding (I1 and I2) and both a 10bp UMI and 5bp monotemplate at the start of both R1 and R2:
10M5S135T8B8B10M5S135T[10M5S+T, 8B, 8B, 10M5S+T]
By utilizing the fgbio toolkit and the Read Structure description, nf-core/fastquorum is able to support the diversity of molecular barcoding schemes. A few that are commercially available are described below:
| Assay | Company | Strand | Randomness | URL |
|---|---|---|---|---|
| SureSelect XT HS | Agilent Technologies | Single | Random | link |
| SureSelect XT HS2 (MBC) | Agilent Technologies | Dual | Random | link |
| TruSight Oncology (TSO) | Illumina | Dual | Nonrandom | link |
| xGen dual index UMI Adapters | Integrated DNA Technologies | Single | Random | link |
| xGen Prism (xGen cfDNA & FFPE DNA Library Prep MC v2 Kit) | Integrated DNA Technologies | Dual | Nonrandom | link |
| NEBNext | New England Biosciences | Single | Random | link |
| AML MRD | TwinStrand Biosciences | Dual | Random | link |
| Mutagenesis | TwinStrand Biosciences | Dual | Random | link |
| UMI Adapter System | Twist Biosciences | Dual | Random | link |
We are working with some of the vendors above to verify that their assays are supported by this pipeline by obtaining test/example data, along with appropriate parameters with which to run this pipeline.
fgbio: the Fulcrum Genomics Bioinformatics toolkit
That brings us to fgbio.
Similar to popular Bioinformatic toolkits samtools, the GATK, and bedtools, fgbio is a collection of command line tools developed by Fulcrum Genomics]fulcrum-genomics-link to analyze primary Genomics data.
Since its conception in 2015, fgbio has been downloaded from Bioconda over three-hundred thousand times, and we use it extensively with our clients at Fulcrum Genomics.


The fgbio toolkit has a wide variety of available tools, with many tools producing tabular quality control metrics.
Particularly relevant for the nf-core/fastquorum pipeline are the tools for working with read-level data containing these unique molecular indexes.
Click the arrows below to expand specific topic areas for the tools:
Tools for working with Unique Molecular Indexes (UMIs, aka Molecular IDs/Barcodes):
- Annotating/Extract UMIs from read-level data:
FastqToBam,AnnotateBamWithUmis,ExtractUmisFromBam, andCopyUmiFromReadName. - Manipulate read-level data containing UMIs:
CorrectUmis,GroupReadsByUmi,CallMolecularConsensusReads,CallDuplexConsensusReads, andFilterConsensusReads. - Collect metrics and review consensus reads:
CollectDuplexSeqMetricsandReviewConsensusVariants.
Tools to manipulate read-level data:
- FASTQ Manipulation:
FastqToBam,ZipperBams, andDemuxFastqs(seefqtk, our rust re-implementation for sample demultiplexing). - Filter, clip, randomize, sort, and update metadata for read-level data:
FilterBam,ClipBam,RandomizeBam,SortBam,SetMateInformationandUpdateReadGroups.
Tools for quality control assessment:
- Detailed substitution error rate evaluation:
ErrorRateByReadPosition. - Sample pooling QC:
EstimatePoolingFractions. - Splice-aware insert size QC for RNA-seq libraries:
EstimateRnaSeqInsertSize.
Tools for adding or manipulating alternate contig names:
- Extract from a NCBI Assembly Report:
CollectAlternateContigNames. - Update contig names in common file formats:
UpdateFastaContigNames,UpdateVcfContigNames,UpdateGffContigNames,UpdateIntervalListContigNames,UpdateDelimitedFileContigNames.
Miscellaneous tools:
- Pick molecular indices (ex. sample barcodes, or molecular indexes):
PickIlluminaIndicesandPickLongIndices. - Find technical/synthetic, or switch-back sequences in read-level data:
FindTechnicalReadsandFindSwitchbackReads. - Make synthetic mixture VCFs:
MakeMixtureVcfandMakeTwoSampleMixtureVcf.
Pipeline Overview
To support the various molecular barcoding schemes, nf-core/fastquorum is organized into two main phases, according to the fgbio best practices: grouping and consensus calling. The pipeline steps for each phase can be best explained with a metro map 🚇:
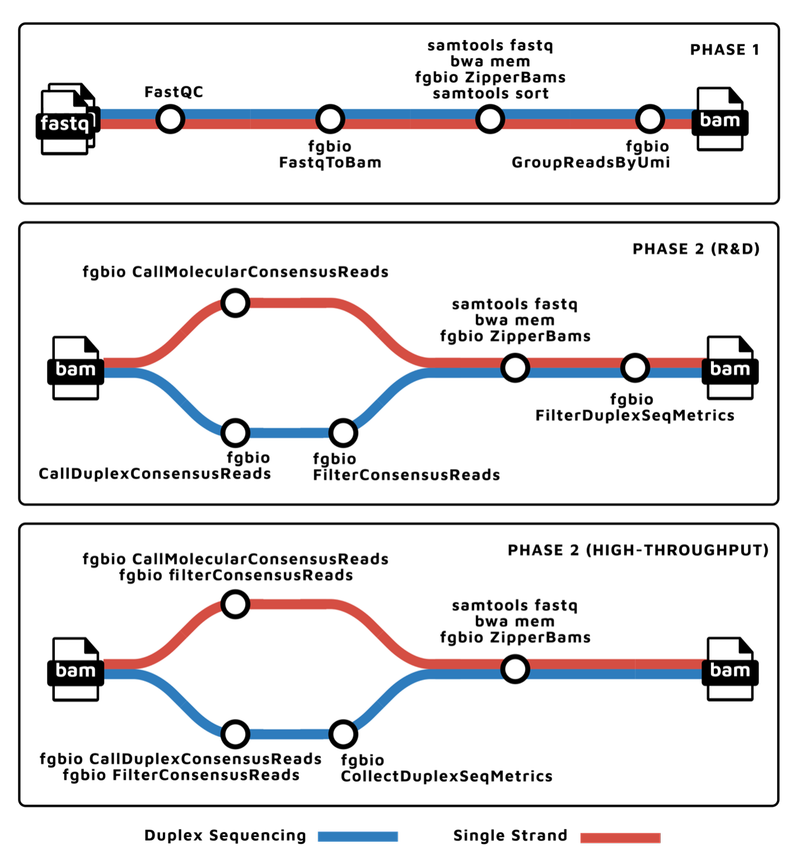
Thank you to James Fellows Yates for these diagrams!
Phase 1: Pre-processing and Grouping
The first phase takes FASTQs as input, and performs the following steps:
- Performs basic Quality Control (with
FASTQC). - Extracts the UMI bases based on the molecular barcoding scheme (with
fgbio FastqToBam). - Aligns the raw reads to the genome (with
bwa,samtools, andfgbio ZipperBams). - Then groups them by genomic coordinate and UMI (with
fgbio GroupReadsByUmi).
This produces the grouped BAM file, where raw reads originating from the same original source molecule are grouped together and tagged.
Phase 2: Consensus Calling
The second phase takes the grouped BAM from phase one, and performs the following steps:
- For each group of raw reads, calls a consensus sequence, thereby eliminating random errors, significantly improving the accuracy of resulting data.
fgbio CallMolecularConsensusReadsis used for single-strand UMI schemes, whilefgbio CallDuplexConsensusReadsis used for duplex sequencing.
- The consensus reads are aligned back to the genome (with
bwa,samtools, andfgbio ZipperBams). - The consensus reads are filtered based on various properties, such as minimum per-molecule or per-base coverage (with
fgbio FilterConsensusReads).
The filtered consensus BAM is ready for downstream analysis, such as variant calling.
Importantly, this R&D version of the second phase allows users to test various tool-level parameters, to optimize them for their data.
The second phase also has a High-Throughput version, for when performance and throughput take precedence over flexibility. This version consensus calls and filters in one step, thereby reducing the number of consensus reads that need to be aligned as well as reducing the number of files that are written to disk.
Release Early, Release Often
… and listen to your nf-core customers.
With the help of some very responsive nf-core maintainers and members, nf-core/fastquorum had its first official release at the Nextflow Summit in Boston (2024).
It supports single-strand and duplex sequencing data, both the R&D and high-throughput fgbio best practices, combining FASTQs across runs and lanes, as well as flexible support for molecular barcoding or UMI schemes.
For more documentation, head over to the nf-core/fastquorum page, visit the fgbio toolkit homepage and fgbio wiki, and join us on our Slack Channel.
It Takes a Nextflow Village
A number of organizations have sponsored and contributed to the development of the fgbio toolkit including Fulcrum Genomics, TwinStrand Biosciences, and Integrated DNA Technologies.
Both Fulcrum Genomics and its current and past team members, along with the nf-core community of maintainers, core team, and contributors have enabled nf-core/fastquorum’s first release.
| Fulcrum Genomics | TwinStrand Biosciences | nf-core Community |
|---|---|---|
| Nils Homer | Michael Hipp | Simon Pearce |
| Tim Fennell | John McGuigan | Adam Talbot |
| Clint Valentine | Thomas Smith | Chad Young |
| Yossi Farjoun | Robert N. Azad | Peter Hickey |
| Jay Carey | Brad Langhorst | |
| Kari Stromhaug | Jordi Camps | |
| Nathan Roach | Brent Pedersen |

If you want to hear this all over again, please check out the recent talk announcing the first release of this pipeline at the Nextflow Summit in Boston (2024):